Newsletter
Cancer treatments using inhibitors of CDK, a cell cycle regulator

Carna Newsletter Vol.16
Cancer treatments using inhibitors of CDK, a cell cycle regulator
(Therapeutic potential of CDK2 inhibitors targeting HR+/HER2- breast cancer with CDK4/6 inhibitor resistance and CCNE1-amplified cancer)
Since the discovery of key regulators of the cell cycle by Dr. Hartwell et al. who were awarded the Nobel Prize in Physiology or Medicine in 2001, research focused on the molecular mechanism of the cell cycle has progressed dramatically.
CDK-cyclin complexes regulate cell cycle progression and are associated with cancer.
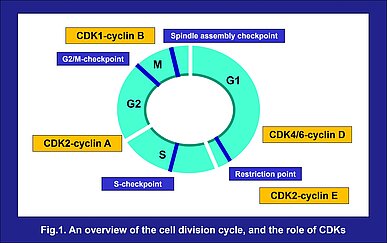
The cell cycle is a fundamental process in life where a series of cellular events occur resulting in the controlled division of one cell and the formation of two identical daughter cells. The cell cycle involves four sequential phases. The S-phase, when DNA replication occurs, and M-phase, when the cell divides into two daughter cells, are separated by gaps known as G1 and G2. Cells prepare for DNA synthesis in G1 and for division in G2. These four phases repeat, resulting in cell proliferation. During the cell cycle, there are several checkpoints which ensure its proper progression, and prevent abnormal progression. The G1/S checkpoint, G2/M checkpoint, and spindle assembly checkpoint, require cyclin-dependent kinases (CDKs) and their cyclins to play the roles of key regulators. The activation of these CDK-cyclin complexes is required to progress through these checkpoints¹⁾ (Fig.1). Deregulation of CDKs has been reported to cause uncontrolled proliferation as well as genomic and chromosomal instability, resulting in human cancers and contributing to cancer progression¹⁾. For their significance, CDKs have always been of interest as drug targets in oncology, and recent advances in understanding have thrust them into the front of active research again.
Cell cycle progression from G1 to S phase
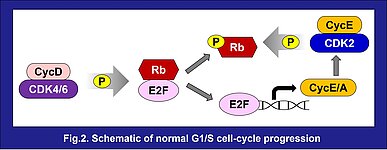
CDK2, CDK4 and CDK6 drive the progression of the cell from G1 to S phase and regulate the cell cycle. When the cell is stimulated by specific mitogens or signaling regulators such as estrogen, the expression of cyclin D is induced. This begins a cascade which initiates changes in gene expression to continue cell cycle progression: Cyclin D forms a complex with CDK4 or CDK6 and then activated CDK4/6-cyclin D complexes phosphorylate and inactivate retinoblastoma protein (Rb), liberating E2F, a transcription factor binding to Rb, to promote transcription of cyclins E and A, which in turn activate CDK2. The activated CDK2-cyclin E complex hyperphosphorylates and inactivates Rb completely, establishing the continued expression of cyclins E and A, as well as other proteins that are essential for S phase²⁾ (Fig.2).
The contribution and challenge of CDK4/6 inhibitors for advanced or metastatic HR+/HER2- breast cancer
Over 70% of breast cancers are estrogen-receptor (ER) positive and human epidermal growth factor 2 (HER2) negative. Endocrine therapy (ET) which blocks estrogen action, is the main current treatment for ER+ breast cancer. However, an estimated 20% of patients have innate resistance to ET, and others acquire resistance to ET over a period of time³⁾. It has been reported that cyclin D1 amplification frequently occurs in ER+ breast cancer⁴⁾ and the maintenance of cyclin D1 expression and Rb phosphorylation – even with ablation of ER activity - was associated with ET-resistance⁵⁾. These findings resulted in CDK4/6 which interacts with cyclin D1, becoming a target of significant interest in the potential treatment for ET-resistance in ER+ breast cancers. Currently developed CDK4/6 inhibitors are able to inhibit Rb phosphorylation, leading to G1 cell cycle arrest in tumor cells, and resulting in inhibition of breast tumor growth. Combinations of CDK4/6 inhibitors + ET resulted in approximately twice as long progression free survival (PFS) time compared to conventional ET therapy only, in patients with advanced or metastatic HR+/HER2- breast cancer, in clinical trials. Currently, combination therapy using CDK4/6 inhibitors with an aromatase inhibitor or fulvestrant (a selective estrogen receptor degrader; SERD) represents the standard of care for advanced or metastatic HR+/HER2- breast cancer⁶⁾⁷⁾.
Resistance against this combination therapy is inevitable and there is currently no clear standard next line treatment, and so, new treatments are desperately needed. Among the diverse mechanisms of CDK4/6 inhibitor resistance reported, aberrant CDK2-cyclin E activity is drawing attention as an associated factor to this resistance⁷⁾. Clinical trials of CDK2 inhibitors are currently underway against HR+/HER2- breast cancer.
Development of CDK2 inhibitors
A secondary analysis of the PALOMA-3 clinical trial of fulvestrant, with or without palbociclib, a selective CDK4/6 inhibitor, in patients with metastatic HR+/HER2- breast cancer, demonstrated that the efficacy of palbociclib was lower in patients with high cyclin E1 mRNA expression⁸⁾. It was also reported that in the palbociclib-resistant HR+ breast cancer cell line, cyclin E protein was significantly overexpressed and phosphorylation of c-MYC, an oncogene, was increased in a CDK2 dependent fashion, and CDK2 inhibitor or CDK2 knockdown inhibited the cell proliferation. Additionally, CDK2 inhibition in combination with CDK4/6 inhibition inhibited cell proliferation even more effectively⁹⁾¹⁰⁾.
CDK2 inhibition causes cells to rapidly lose substrate phosphorylation, but it rebounds within several hours. It is suggested that CDK4/6 activity ‘backstops’ inhibition of CDK2 and sustains the proliferative program by maintaining Rb hyperphosphorylation, active E2F transcription, and cyclin A2 expression, enabling re-activation of CDK2 in the presence of drug²⁾. This suggests that simultaneous combination of CDK2 and CDK4/6 inhibitors or an inhibitor targeting CDK2/4/6 collectively, such as RGT-419B are potential therapeutic candidates to overcome CDK4/6 inhibitor-resistant HR+/HER2- breast cancer.
CDK2 inhibitor as a potential treatment for CCNE1-amplified cancers.
Amplification of CCNE1, a cyclin E1 coding gene, was identified in approximately 20% of ovarian carcinomas and it was found the presence of CCNE1 amplification correlated with shorter disease-free survival, and overall survival, after standard chemotherapy¹¹⁾. Therefore, CDK2 is an attractive target in treating CCNE1-amplified ovarian cancer due to its relative specificity for cyclin E and the essential role CDK2 plays in the activated CDK2-cyclin E1 complex. The use of a selective CDK2 inhibitor or CDK2 knockdown inhibited proliferation of CCNE1-amplified ovarian cancer cells¹⁰⁾¹²⁾. In a CCNE1-amplified ovarian tumor model, a selective CDK2 inhibitor showed antitumor activity and additionally, the combination of a CDK2 inhibitor with chemotherapy including carboplatin, the standard of care, showed more effective antitumor activity and induced tumor regression¹³⁾. CCNE1 amplification is prevalent across various tumor types, with ovarian cancer among them¹³⁾. Therefore, agents targeting CDK2 have the potential to become an important therapeutic option for diverse cancers in the future.
CDK products & services from Carna to support inhibitor development.
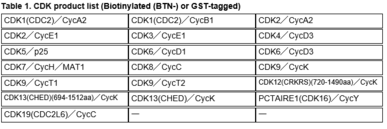
Kinase Proteins (Table 1)
All of Carna’s CDK kinase proteins shown below are manufactured in-house, and available with a GST tag, as well as with a single biotin-tag at the protein’s N-terminus. These proteins are useful in various biochemical assays, including kinase activity and inhibitor binding assays.
Also Available are:
・Kinase Assay Kits (for MSA, FP(IMAP™) or ELISA)
・Kinome Wide Activity-based Biochemical Screening/Profiling Assay Services
・QuickScout® Cell Cycle panel Biochemical Assay Services using 30 relevant kinases involved in cell-cycle regulation
・Kinome -wide NanoBRET™ TE Intracellular Kinase Assay Services
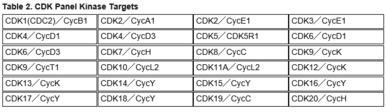
・CDK Panel Assay Services using the 24 fixed CDK targets shown in Table 2.
Or use both our biochemical and cell-based assay services to best evaluate your compounds.
If you are interested in our products or services, please feel welcome to contact us at info@kinaselogistics.com.
References:
- ) J Med Chem. 2019; 62(9):4233-4251. Tadesse S.
- ) Cell. 2023; 186(12):2628-2643.e21. Arora M.
- ) Oncotarget. 2018; 9(91): 36252-36253. Anurag M.
- ) Nature. 2012; 490(7418):61-70. Cancer Genome Atlas Network.
- ) Endocr Relat Cancer. 2011; 18(3):333-45. Thangavel C.
- ) Int J Mol Med. 2022; 50(4):128. Huang J.
- ) Front Cell Dev Biol. 2023; 11:1148792. Zhou FH.
- ) J Clin Oncol. 2019; 37(14):1169-1178. Turner NC.
- ) Cancers (Basel). 2020; 12(12):3566. Pandey K.
- ) Incyclix poster, Cancer Res. 2023; 83 (7_Supplement): 5994. Trub AG.
- ) Cancer. 2010; 116(11):2621-34. Nakayama N.
- ) Clin Cancer Res. 2013; 19(21):5960-71. Etemadmoghadam D.
- ) Blueprint Medicines poster, AACR Annual Meeting 2022; 2306. Brown V.
Further news
-
 Newsletter
Newsletter
ALK drug resistant mutations: Challenges for the treatment of lung cancer
Read moreCarna Newsletter Vol.15
-
 Offers
Offers
2023 Year-End Campaign
Read moreWe’ve got good news for you: a special discount for human Kinases from Carna Biosciences.
-
 Technical Note
Technical Note
DGKα and DGKζ are key targets for cancer immunotherapy
Read moreCarna Newsletter Vol.14